Dexmedetomidine attenuates subarachnoid hemorrhage-induced acute lung injury through regulating autophagy and TLR/NFκB signaling pathway
Article information

Abstract
Background
Acute lung injury (ALI) is the most serious complication of subarachnoid hemorrhage (SAH). We investigated role of autophagy and inflammatory signaling pathways in lung damage and therapeutic effects of dexmedetomidine (DEX).
Methods
Fifty male Wistar rats were randomly divided into five groups: sham, SAH, SAH+ DEX5, SAH+DEX25, and SAH+DEX50. SAH was induced using endovascular perforation technique. All rats received mechanical ventilation for 60 min. At 2 and 24 h of SAH induction, SAH+DEX groups were treated with 5, 25, and 50 µg/kg of DEX, respectively. Histological ALI score and pulmonary edema were assessed after 48 h. Lung expression of LC3B, ATG3, p62, TLR4, TLR9, and NFκB was assessed using western blotting and quantitative PCR. Blood levels of IL-6, IL-1β, IFN-γ, and TNFα were also assessed.
Results
SAH induced ALI and pulmonary edema, which were attenuated in SAH+DEX5 (P < 0.001 for both) and SAH+DEX25 groups (P = 0.001 and P < 0.001 for ALI and edema, respectively). Lung expressions of LC3B and ATG3 were upregulated in SAH group, which was attenuated in SAH+DEX5 and SAH+DEX25 groups. Lung expressions of TLR4, TLR9, and NFκB were increased in SAH group, which was attenuated in SAH+DEX5 group. Blood IL-6 level was increased in SAH group and attenuated in SAH+DEX5 and SAH+DEX25 groups. Blood IFN-γ level was lower in SAH group than in sham group, and it was increased in SAH+DEX25 group.
Conclusions
Low-dose DEX treatment after SAH may protect against ALI by disrupting pathological brain-lung crosstalk and alleviating autophagy flux and TLR-dependent inflammatory pathways.
Introduction
Subarachnoid hemorrhage (SAH), a leading cause of death and neurological devastation, mostly induces extracerebral organ dysfunction. The lungs are most commonly affected as they are particularly vulnerable to harmful signals, such as massive sympathetic stimulation and systemic inflammatory responses, which are activated during SAH [1]. Once attacked, the lungs become highly sensitive to mechanical ventilation, surgery, and infection that follow after severe brain injury and get irreversibly damaged [2]. Pulmonary complications are largely responsible for the mortality and healthcare burden of SAH [1]. The current therapeutic approach is mainly supportive, since the exact molecular mechanism of SAH-induced acute lung injury (ALI) has not been fully elucidated.
Autophagy maintains cellular homeostasis by recycling damaged materials and regulating cellular bioenergetics [3]. Accordingly, coping with various cellular stresses depends on its activity [4]. As autophagy balances the beneficial and detrimental actions of immunity, the pathogenesis and prognosis of inflammatory diseases are strongly affected by its involvement [5]. The implication of autophagy in lung injury is complex because its function is multifaceted according to the cell types and environment [6], which complicates autophagy-based diagnostic and therapeutic approaches. Likewise, a number of previous experimental studies on ALI models support autophagy activation as a mechanism of action for several therapeutic agents, while a few have reported that attenuating autophagic activity facilitates protection against damage [7]. To date, molecular switches between the injurious or protective activity of autophagy in the lungs after SAH have not been explored. The association of autophagy with pro-inflammatory signaling pathways involved in brain-lung crosstalk remains elusive.
Dexmedetomidine (DEX), a highly selective Alpha 2-adrenergic receptor agonist, is a widely used sedative and analgesic agent. A large body of evidence supports its organ-protective effects, including anti-inflammatory, antioxidative, and sympatholytic properties [8]. Regarding its impact on the lungs, a number of experimental studies have revealed that DEX attenuates various types of ALI [8], which is consistent with promising clinical results in patients with pulmonary dysfunction and those undergoing lung surgery [9,10]. Recently, autophagy suppression has been suggested as a mechanism of lung protection by DEX in ALI induced by ischemia-reperfusion, lipopolysaccharide instillation, and toxic shock [11–13]. However, there are reports that autophagy activation induced by DEX plays a central role in protection against neuronal and myocardial damage [14,15].
In this study, we investigated whether autophagy promotes or protects against ALI induced by SAH in a rat model of endovascular perforation undergoing mechanical ventilation. We also evaluated the protective effects of DEX against SAH-induced ALI with the mechanistic insights.
Materials and Methods
Ethical statement and animal preparation
The experimental protocol was approved by our institutional animal care and use committee (No. 2015-0217). All experimental animals were handled in accordance with the National Institute of Health (NIH) guidelines and the Guide for the Care and Use of Laboratory Animals. Fifty male Wistar rats aged 20–22 weeks were housed in temperature- and humidity-controlled animal quarters with a 12 h light/dark cycle and free access to food and water before the experiments.
SAH model
SAH was induced using the endovascular perforation technique, as previously described [16]. Briefly, after rats were anesthetized with 3% isoflurane in 50/50% medical air/oxygen, the left common carotid artery (CCA) and its branches were exposed. Two small loose ligatures were placed on the left external carotid artery (ECA) and a small distal incision was made. A 3-0 monofilament nylon suture was inserted through this incision, and the loose ligatures were fastened over the filament when advanced to the internal carotid artery (ICA). The filament was continuously fed into the vessel until resistance was encountered, indicating that the filament had reached the intracranial bifurcation of the ICA. The filament was advanced further by approximately 3 mm to perforate the artery, and was then immediately withdrawn. In the sham group, sutures were inserted into the ECA, but arterial perforation was not performed. The incision was closed after surgery.
Experiments continued only on the rats that developed a considerable amount of hemorrhage; the severity of SAH was assessed after perfusion by a blinded observer based on a previously described grading system [17]. Briefly, the brain base was divided into six parts, each of which was scored from 0 to 3 points according to the amount of blood extravasated into the subarachnoid space. The total score was defined as the sum of six segments, ranging from 0 (no hemorrhage) to 18 (most severe hemorrhage). SAH rats with a score ≤ 7 were excluded from the study. No hemorrhaging was observed in the sham-operated rats and thus received a score of 0.
Mechanical ventilation and drug administration
Rats were randomly assigned to the following five groups: sham, SAH, SAH+DEX5, SAH+DEX25, and SAH+DEX50 (n = 10 each). Immediately after SAH or sham surgery, all rats were intubated and mechanically ventilated for 60 min in a volume-controlled mode with a tidal volume of 6 ml/kg, at a fractional inspired oxygen concentration of 0.5, inspiration to expiration ratio of 1 : 1, and respiratory rate of 80 breaths/min. A 1.5% isoflurane anesthesia was continued during mechanical ventilation. Two and 24 h after surgery, the sham and SAH groups were intraperitoneally injected with 2 ml of normal saline, while the SAH+DEX5, SAH+DEX25, and SAH+DEX50 groups received the same volumes of 5, 25, and 50 μg/kg of DEX (Precedex®, Hospira Inc., USA) intraperitoneally.
Neurobehavioral assessment
The neurobehavior of the rats was evaluated (n = 7–10 per group) 48 h after surgery using the previously described modified limb-placing test (MLPT) [18]. Ambulation was assessed by walking with the lower limbs, which was graded as follows: 0 points (normal walking), 1 point (mildly ataxic gait with flat toes under the body), 2 points (ataxia with knuckle walking), 3 points (presence of lower limb movement but unable to walk), and 4 points (absence of lower limb movement, dragging legs). Second, the placing/stepping reflex was assessed by dragging the dorsum of the hind limb over the edge of the table, which induced lifting and placing responses such as stepping, and graded as follows: 0 points (well-coordinated lifting and placing response), 1 point (mildly uncoordinated and weakened response), 2 points (severely impaired response or knuckle walking), and 3 points (no response). The sum of the scores in the two tests (ranges 0–7) was defined as the motor deficit index (MDI). Rats with an MDI ≥ 3 were considered paraplegic.
Lung histopathology and immunohistochemistry
The rats (n = 7–10 per group) were anesthetized with isoflurane 48 h after SAH or sham surgery and intracardially perfused with 1% phosphate-buffered saline (PBS) immediately after blood collection. The lower lobe of the left lung was harvested, fixed in 4% paraformaldehyde for 24 h, and then immersed in 30% sucrose overnight. The tissue was embedded in paraffin and cut into 4 μm sections. For histopathological scoring and immunohistochemistry, tissue sections were deparaffinized, rehydrated using 10 mM citrate buffer (pH 6.0) in a microwave at 750 W for 20 min, and subsequently cooled. Half of the sections were stained with hematoxylin and eosin (H&E) and scanned under light microscopy by two experienced pathologists who were blinded to the group assignment. Alveolar congestion, hemorrhage, alveolar wall thickness, and inflammatory cell aggregation were evaluated according to previously described criteria [4], with some modifications: 0, normal tissue; 1, minimal inflammatory change; 2, slightly thickened alveolar septa by mild infiltration of inflammatory cells, but without obvious architectural distortion; 3, formation of nodules and/or severe inflammatory changes that distort the architecture; and 4, total fibrous obliteration of the field.
The other half of the sections was incubated in 3% H2O2 in PBS at room temperature for 10 min and then washed with 1X PBS. The samples were blocked with REAL Peroxidase-Blocking Solution (S2023, DAKO, Denmark) for 20 min, incubated with LC3B (1 : 1000, Novus Biologicals, USA) primary antibody overnight at 4°C, and then washed with 1X PBS. The samples were subjected to the REAL EnVision Detection System (DAKO K5007), washed thoroughly under tap water, and mounted on slides. Mayer’s hematoxylin was used for counterstaining. Images of the colonies were captured using a light microscope OLYMPUS BX40 (40× magnification).
Lung wet/dry weight ratio
To assess the development of pulmonary edema, the lung wet/dry weight ratio was measured in the fresh right lung (n = 7–10 each), as described previously [19]. Briefly, lung samples were extracted immediately after perfusion, and excess fluid was blotted. The samples were then placed in pre-weighed glass vials and weighed (wet weight). The samples then were wrapped in tinfoil and placed in an oven at 80°C for 48 h. The wet/dry weight ratio was calculated as [(wet weight − dry weight) / wet weight] × 100.
Since lung histology and the degree of pulmonary edema in the SAH+DEX50 group did not show any beneficial effects of the agent, this group was excluded from further experiments for mechanistic analysis.
Quantitative real-time polymerase chain reaction (PCR)
Immediately after intracardiac perfusion, tissue samples from the left upper lobe were freeze-clamped in liquid nitrogen and stored at −80°C. Total RNA was isolated using an RNeasy Mini Kit (Qiagen, USA). Complementary DNA (cDNA) was synthesized from 1 µg of total RNA using AccuPower RT premix kits (BIONEER, Korea). Real-time PCR analysis was performed using the TB GreenTM Premix Ex Taq II kit (TAKARA, Japan) and QuantStudio3 (Applied Biosystems, USA) according to the manufacturer’s instructions. Each sample was analyzed in quadruplicate, and target genes were normalized to the reference housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Fold differences were calculated for each group using the normalized CT values for the sham groups. The primer sequences for real-time PCR are as follows: LC3B: Forward 5’-AGAGCGATACAAGGGTGAGA-3’, Reverse 5’-ACTTCAGAGATGGGTGTGGA-3’; TLR 4: Forward 5’-CCAGAGCCGTTGGTGTAT CT-3’, Reverse 5’-TACAATTCGACCTGCTGCCT-3’; TLR 9: Forward 5’-AAATCGTTCAGTGAGC TCCC-3’, Reverse 5’-CTGAAGTTGTGGCCTATCCC-3’; GAPDH: Forward 5’-CTGAGAATGGGAAGCTGGTC-3’, Reverse 5’-CTCCACGACATACTCAGCAC-3’.
Western blotting
Immediately after intracardiac perfusion, tissue samples from the left upper lobe were freeze-clamped in liquid nitrogen and stored at −80°C. Frozen tissues were homogenized in western lysis buffer (4°C) containing 20 mM Tris, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid, 1 mM ethylene glycol tetra acetic acid, 1% Triton X-100, complete protease inhibitor cocktail (one tablet per 10 ml; Roche Diagnostics Corporation, Indianapolis, IN), and phosphate inhibitor cocktail set II (EMD Biosciences, Germany). Thereafter, the homogenates were centrifuged at 12,000 rpm for 30 min at 4°C. The total protein concentration was determined using the Quick Start Bradford reagent (Bio-Rad, USA), with bovine serum albumin as a standard. Whole-cell protein extracts (50 µg) were denatured by heating to 100°C for 5 min, separated on 10–15% sodium dodecyl sulfate-polyacrylamide electrophoresis (SDS-PAGE) gels, and transferred onto Immobilon-P transfer membranes (Millipore, USA). The membranes were incubated successively with ATG3 (1:1000, Cell Signaling Technologies, USA), p62 (1:1000, Abnova, Taiwan), LC3B (1:2000, Novus Biologicals, USA), Toll-like receptor (TLR) 4 (1:200, Santa Cruz Biotechnology, USA), TLR 9 (1:200, Santa Cruz Biotechnology), phospho-nuclear factor-kappaB (NF-κB) (1:200, Santa Cruz Biotechnology), NF-κB (1:1000, Cell Signaling Technologies), or GAPDH (1:1000, Cell Signaling Technologies) overnight at 4°C. The signal was detected using WestGlow FEMTO chemiluminescent substrate (BIOMAX, Korea). GAPDH was used as the protein-loading control.
Enzyme-linked immunosorbent assay (ELISA)
Levels of IL-1β (R&D Systems, USA), TNF-α (Abcam, UK), IL6 (BioLegend, USA), and IFN-γ (MyBioSource, USA) in the serum were determined using ELISA commercial kits according to the manufacturer’s instructions.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 9 software (GraphPad Software, Inc., USA) and SPSS version 23 (IBM Corp., USA). Statistical significance of comparisons among the groups was analyzed using one-way analysis of variance (ANOVA) followed by Bonferroni post-hoc analysis for multiple comparisons. All data are presented as mean ± SD or standard error of the mean (SEM). Statistical significance was defined as 2-sided P < 0.05.
Results
Effects of DEX on SAH grade and neurological deficit
Mortality before completion of the experimental protocol was 30% and 50% in the SAH group and the SAH+DEX50 group, respectively. Two rats in the SAH+DEX5 group and three in the SAH+DEX25 group (out of 10 rats in both groups) showed SAH grading scores of ≤ 7 and were excluded from the study. The scores in the remaining rats with SAH induction were similar among the groups. In the neurobehavioral assessment (Table 1), the SAH group showed severe neurological deficit, contrary to the sham group with normal neurobehavior (3.7 ± 0.9 vs. 0.1 ± 0.4, P < 0.001). However, the SAH+DEX50 group (5.4 ± 0.7) had significantly higher deficit scores than the other groups (P < 0.001 vs. sham group, SAH+DEX5 group, and SAH+DEX25 group and P < 0.001 vs. SAH group). Nine out of ten rats in the SAH were paraplegic (MDI ≥ 3), whereas none of the sham group, SAH+DEX5, and SAH+DEX25 groups developed paraplegia. All eight rats in the SAH+DEX50 group developed paraplegia.

Neurobehavior Outcomes Assessed by MLPT
Effects of DEX on severity of ALI and pulmonary edema
As shown in Fig. 1A, H&E staining of lung tissue in the SAH group revealed thickened and broken alveolar wall, inflammatory cells infiltration in alveoli, and patchy hemorrhage with fibrinous exudate in the alveola and interstitium, in contrast with that in the sham group showing normal lung architecture and no or minimal inflammation, which was probably induced by mechanical ventilation. Pathological changes were ameliorated by treatment with 5 µg/kg (SAH+DEX5 group) and 25 µg/kg (SAH+DEX25 group) DEX. However, histology of the SAH+DEX50 group showed severe injury, diffuse inflammatory cell infiltration in the alveoli and interstitium, and thickened or disrupted alveolar walls. Quantification of the injury demonstrated a significant attenuation of the ALI by treatment with 5 µg/kg and 25 µg/kg DEX (1.5 ± 0.5 vs. 3.2 ± 0.9 in SAH+DEX5 group and SAH group, P < 0.001; and 1.7 ± 0.5 vs. 3.2 ± 0.9 in SAH+DEX25 and SAH groups, P = 0.001; Fig. 1B). The ALI score in the SAH+DEX50 group was higher than those in the SAH+DEX5 and SAH+DEX 25 groups, but the difference was not statistically significant.
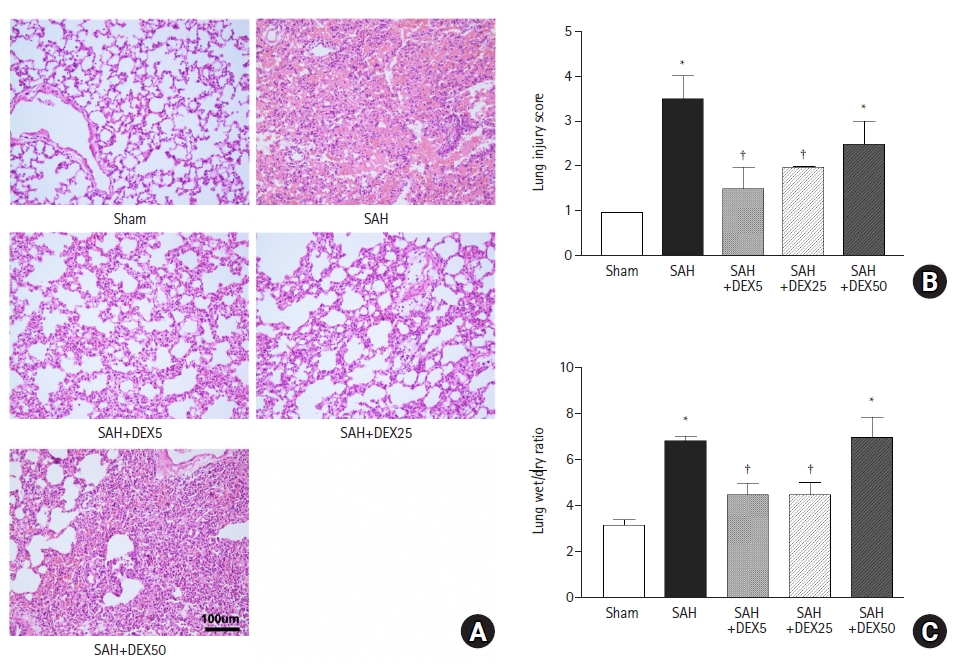
Effects of DEX on severity of lung injury and pulmonary edema. (A) Representative histology images of lung tissue assessed by hematoxylin–eosin staining at 48 h after SAH induction. (B) A quantification of severity of lung injury assessed by modified ALI score that ranges from 0 to 4. (C) A quantification of pulmonary edema assessed by ratio of lung wet/dry weight. n = 7–10 per group and three technical replicates per each rat. Values are presented as means and SD. *P < 0.05 vs. sham group, †P < 0.05 vs. SAH group. ALI: acute lung injury, SAH: subarachnoid hemorrhage, DEX5, 25, 50: dexmedetomidine 5, 25, 50 µg/kg administered at 2 and 24 h after surgery.
The lung water content, assessed by wet/dry weight ratio, was significantly increased in the SAH group compared with the sham group (6.4 ± 0.6 vs. 3.1 ± 0.3, P < 0.001; Fig. 1C). The SAH+DEX5 group and the SAH+DEX25 group showed lower lung wet/dry ratio compared with the SAH group (4.5 ± 0.5 and 4.1 ± 0.4 vs. 6.4 ± 0.6, P < 0.001, respectively). The wet/dry ratio in the SAH+DEX50 group was comparable to that in the SAH group (6.9 ± 0.9 vs. 6.4 ± 0.6, P = 0.794) and was significantly higher than that in the SAH+DEX5 and SAH+DEX25 groups (both P < 0.001).
Effects of DEX on expression of autophagy-regulatory factors in lung tissue
To determine the role of autophagic activation in SAH-induced ALI and the effects of DEX, we evaluated the gene and protein expressions of essential regulatory factors for autophagy (Fig. 2).
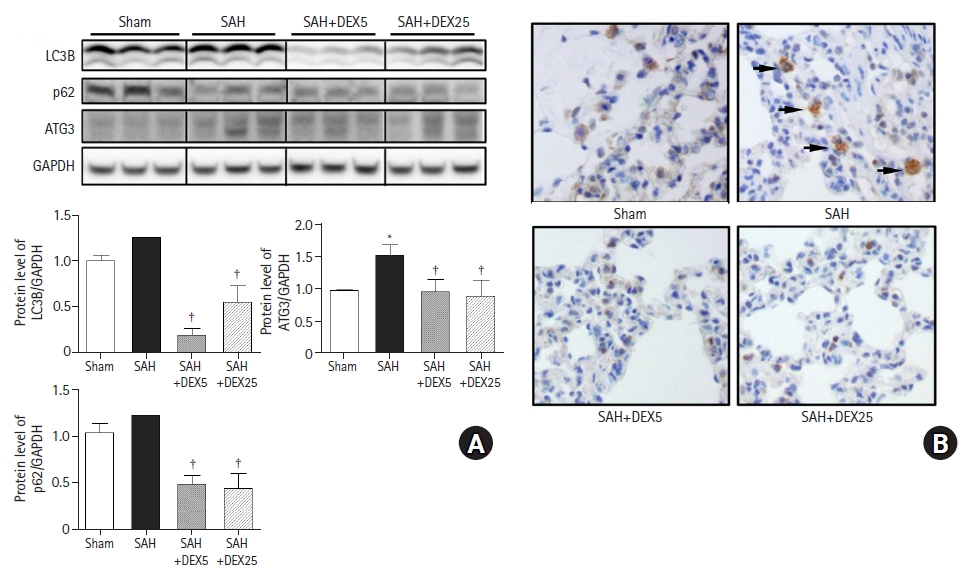
Effects of DEX on expression of autophagy-regulatory factors in lung tissue. (A) Representative western blots and a quantification of protein levels of LC3B, ATG3, and p62 normalized to GAPDH. n = 7–10 per group and three technical replicates per each tissue sample. (B) Representative LC3B immunohistochemistry images. Black arrows indicate LC3B-positive cells (brown color). Values are presented as means and SD. *P < 0.05 vs. sham group, †P < 0.05 vs. SAH group. SAH: subarachnoid hemorrhage, DEX5, 25: dexmedetomidine 5, 25 µg/kg administered at 2 and 24 h after surgery.
The protein expression levels of LC3B and p62 between the sham and SAH groups were not significantly different (P = 0.096 and 0.065, respectively) compared to the sham group, while the level of ATG3 was significantly higher in the SAH group than in the sham group (Fig. 2A). The levels of LC3B and ATG3 in the SAH+DEX5 and SAH+DEX25 groups were significantly lower than those in the SAH group. The mRNA expression of LC3B was significantly increased in the SAH group compared with the sham group, while the expression was significantly decreased in the SAH+DEX5 group compared with the SAH group (data not shown). As shown in Fig. 2B, there were a greater number of LC3B positive cells in the tissue sections of the SAH group than in those of the sham group. The SAH+DEX5 and SAH+DEX25 groups showed lower expression of LC3B positive cells than the SAH group.
Effects of DEX on expression of TLRs and NFκB in lung tissue
To evaluate whether TLR signaling pathways are responsible for lung inflammation induced by SAH and the effects of DEX, we assessed the lung expression levels of TLR4, TLR9, and downstream molecules (Fig. 3). The protein and mRNA expression levels of TLR4 and TLR9 were significantly higher in the SAH group than those in the sham group. The mRNA expression of TLR4 was significantly lower in the SAH+DEX5 and SAH+DEX25 groups than in the SAH group, but the decrease in protein expression levels was not statistically significant. The protein and mRNA expression levels of TLR9 were lower in the SAH+DEX5 group compared with the SAH group. The protein expression level of phosphorylated NFκB was significantly higher in the SAH group than in the sham group, while the levels were significantly decreased in the SAH+DEX5 group. In the additional qPCR analysis, mRNA levels of IL-6 and IL-18 were significantly higher in the SAH group than in the sham group, while the levels in the SAH+DEX5 and SAH+DEX25 groups were significantly lower than those in the SAH group (data not shown).
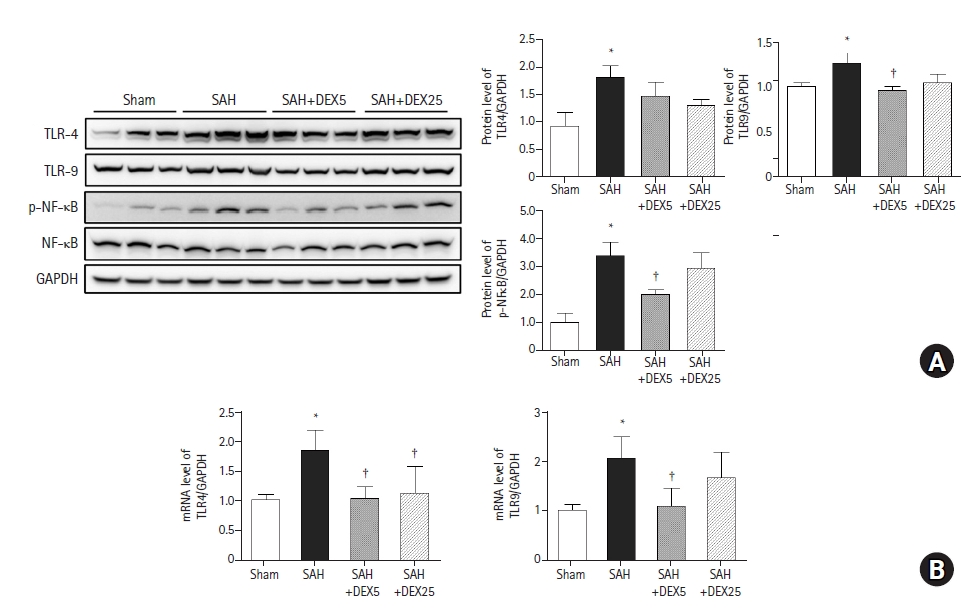
Effects of DEX on expression of TLRs and NFκB in lung tissue. (A) Representative western blots and a quantification of protein levels of TLR4, and TLR9 normalized to GAPDH and phosphorylated NFκB normalized to NFκB. (B) A quantification of mRNA levels of TLR4 and TLR9 normalized to GAPDH assessed by quantitative PCR. n = 7–10 per group and three technical replicates per each tissue sample. Values are presented as means and SD. *P < 0.05 vs. sham group, †P < 0.05 vs. SAH group. SAH: subarachnoid hemorrhage, DEX5, 25: dexmedetomidine 5, 25 µg/kg administered at 2 and 24 h after surgery.
Effects of DEX on systemic levels of inflammatory cytokines
In the ELISA to determine the role of systemic inflammation in mediating the brain-lung crosstalk (Fig. 4), the blood level of IL-6 was significantly higher in the SAH group than in the sham group, and the level was significantly decreased in the SAH+DEX5 group and in the SAH+DEX25 group, compared with the SAH group. The blood level of IFN-γ was significantly lower in the SAH group than in the sham group, and the level was significantly higher in the SAH+DEX25 group than in the SAH group. Blood levels of IL-1β and TNF-α did not differ among the groups.
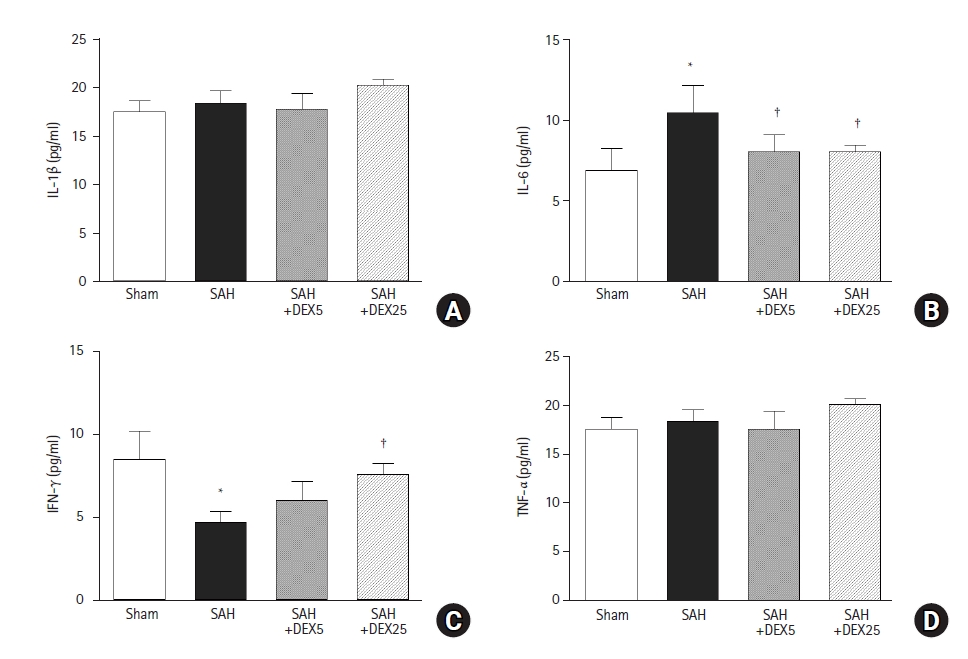
Effects of DEX on circulating levels of inflammatory cytokines. (A-D) A quantification of blood serum levels of IL-1β, IL-6, IFN-γ, and TNF-α. n = 7–10 per group and three technical replicates per each tissue sample. Values are presented as means and SD. *P < 0.05 vs. sham group, †P < 0.05 vs. SAH group. SAH: subarachnoid hemorrhage, DEX5, 25: dexmedetomidine 5, 25 µg/kg administered at 2 and 24 h after surgery.
Discussion
Autophagy has recently emerged as an adaptive response to pulmonary insults induced by various stresses that manifest as chronic obstructive pulmonary disease, asthma, pulmonary fibrosis, and acute respiratory distress syndrome (ARDS) [7]. One of the most important autophagic activities in this regard is the regulation of inflammation [20]. Considering that SAH frequently causes ALI, which is characterized by an inflammatory process in lung tissue, autophagy might play an important role in the development of SAH-induced lung damage, which has not yet been revealed. In this study on the SAH rat model, we found that the crucial regulators of autophagy were significantly altered towards the activation of autophagy, which was linked to pulmonary edema, alveolar infiltration of inflammatory cells, and architectural distortion. This finding suggests that autophagic flux may be responsible for pulmonary complications in patients with SAH. Furthermore, we observed that DEX treatment after SAH reversed lung injury and pulmonary edema with attenuation of increases in the tissue expression of autophagy regulators, which confirms that autophagy might serve as a potential therapeutic target in SAH-induced ALI.
Pattern-recognition receptors (PRRs) that are activated by danger-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns were demonstrated to promote activation of autophagy in diverse inflammatory states [5]. TLR4, one of the best-characterized PRRs, is known to specifically recognize bacterial lipopolysaccharides (LPS) as well as several DAMPs, especially high-mobility group box (HMGB)-1, and activates NFκB and mitogen-activated protein kinases (MAPK), thereby leading to lung inflammation either by various direct or indirect insults [21,22]. Our study is the first to reveal that activation of TLR4 and NFκB in lung tissues after SAH could be induced by the massive release of HMGB-1 from neuronal cells [23]. TLR4 has been demonstrated to trigger the activation of autophagy via a number of downstream signaling pathways, including TNF receptor-associated factor (TRAF)6, Toll/IL-1 receptor domain-containing adaptor interferon-B (TRIF), phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway, and the MyD88/NFκB pathway [24]. A previous in vitro study on macrophages identified TLR4 as a major trigger for LPS-induced proinflammatory cytokine release and autophagic activation [25]. In contrast, mediators of autophagy have also been demonstrated to stimulate TLR4 and NF-κB, thereby exaggerating inflammation in various diseases [26]. In terms of lung injury, a previous study on a minipig model of lung ischemia-reperfusion (IR) demonstrated that autophagy amplifies lung inflammation by activating TRAF6, MAPK, and NF-κB [27]. The role of autophagy in aggravating the inflammatory response and lung damage via mTOR signaling has also been demonstrated in LPS-induced ALI [28]. Autophagic activation in the lung tissues of SAH rats along with the severe inflammatory changes and upregulated genes encoding IL-6 and IL-18 in our study may support the interactive role of autophagy and inflammatory pathways initiated by TLR4 in pathological brain-lung crosstalk.
In contrast, a number of reports have suggested a protective role of autophagic activation against ALI. A study on an LPS-induced ALI mouse model revealed the cytoprotective effects of autophagy mediated by endoplasmic reticulum stress and exacerbation of cytotoxicity by administering an autophagy inhibitor [29]. A study on a traumatic brain injury (TBI) rat model showed that TBI promoted autophagy flux in lung tissues, which was consistent with our findings; however, enhanced autophagy flux attenuated lung inflammation and apoptosis, while inhibition of autophagy flux exacerbated lung damage [30]. The double-edged sword of autophagy modulation in cell survival, inflammation, and immunity, due to highly variable autophagy function according to cell type and causative stimuli, has been widely recognized [26]. Although SAH-induced ALI may share some pathophysiology of brain-lung crosstalk with TBI-induced ALI, a difference may also exist, which can be inferred by distinct proteins in cerebrospinal fluid (CSF) between TBI patients and those with non-TBI, including SAH [31]. In addition, superimposed ventilation-induced lung injury (VILI) in the current study, but not in the TBI model [30], could have contributed to the discrepancy in the role of autophagy. VILI is characterized by increased pulmonary vascular permeability, infiltration of immune cells, and robust activation of pro-inflammatory signaling and is widely recognized to aggravate lung damage in patients with ALI/ARDS [32]. Autophagy has been implicated in inflammatory injury during mechanical ventilation via inflammasome activation and stimulation of the NFκB pathway [33,34]. Increased lung expression of TLR9 in SAH rats in the current study may support a recent report on the role of TLR9 in mechanical ventilation-induced inflammatory injury through the MyD88/NFκB pathway [35]. Synergistic interactions between autophagy and the TLR9/NFκB signaling pathway may potentially contribute to double-hit lung injury, which requires further investigation.
Given the safety and efficacy profile and widely recognized lung-protective properties of DEX [8], we investigated its therapeutic effects on SAH-induced ALI. Although autophagic function in the role of DEX in protecting organs has been controversial, recent evidence from experimental studies on ALI supports that DEX may inhibit activation of autophagy in injured lung tissues, which is consistent with our findings. In a rat model of toxic shock-induced ALI, a single dose of 50 µg/kg DEX downregulated lung expression of LC3B and Beclin-1, as well as inflammatory proteins, with attenuation of pulmonary edema and apoptosis [13]. A study on a rat model of lung IR injury also revealed that pre-treatment with 1–10 µg/kg DEX prevented ALI along with the downregulation of BCL2 Interacting Protein3 (BNIP3), a potent inducer of mitochondrial autophagy, as well as LC3B [11]. The mechanisms for the inhibition of autophagy may include the potent anti-inflammatory property of DEX. In a TBI-induced ALI mouse model, a single dose of 50 µg/kg DEX exerted protective effects via suppression of HMGB-1 signal transduction in the lung tissue [36]. A study in a rat model of sepsis-induced ALI revealed that treatment with 10–20 µg/kg DEX inhibited lung inflammation by suppressing the TLR4/MyD88/NFκB [37]. These results are consistent with our finding of attenuated upregulation of TLRs, NF-κB, and ILs by DEX along with autophagy inhibition, which may indicate an interrupted vicious cycle of autophagic damage and inflammation in SAH-induced ALI.
A dosing protocol used in our experiment was based on previous studies that showed protective effects described above. We also selected repeating dose to reflect the clinical situation of SAH management better. In contrast to the single dose of 50 µg/kg that showed protective effects against the secondary ALI previously [13,36], a double administration of 50 µg/kg at 2 h and 24 h after brain damage in the current study did not bring any benefits. Considering the poor neurobehavioral scores in this group, we could assume that repeated high dose could have led to exaggerated pathological activation of brain-lung crosstalk and consequent severe lung damage. Indeed, there is a report on neurotoxicity of 100 µg/kg DEX, although the toxic dose of this agent is still controversial [38,39]. The timing of drug administration in the current study, which occurred after brain insults could also be responsible for neurotoxicity because the brain may become more vulnerable to toxicity compared to pre-treatment or preconditioning. The second-hit damage by mechanical ventilation in our study compared with those studies without it could be another reason of the different result. A high dose of DEX may also be ineffective or harmful to the lung itself, although there is scarce data to support this hypothesis. One previous study that evaluated the effects of 100 µg/kg on ALI and reported no benefit, while the lower doses, 10 µg/kg and 50 µg/kg, exerted significant protection [40]. Notably, the protective effects observed in our SAH+DEX5 and SAH+DEX25 groups were comparable and not dose-dependent. Moreover, tissue expression of TLR9 and NF-κB was only attenuated in the SAH+DEX5 group, and the level of LC3B in the SAH+DEX5 group was significantly lower than that in the SAH+DEX25 group. These findings suggest that the dosing protocol of DEX post-SAH should be very cautious, and a more sophisticated evaluation needs to be performed.
Although inflammation is a widely accepted mechanism of brain-lung crosstalk, data on the circulating levels of inflammatory mediators are scarce. Remarkable changes in blood IL-6 levels according to the occurrence of SAH and treatment with DEX, which were associated with ALI severity in the current study may indicate systemic inflammation as a connecting link between the brain and lungs. We previously observed in our rat model of SAH-induced vasospasm that blood and CSF levels of IL-6 were strongly correlated with the severity of vasospasm and the therapeutic effects of DEX [41], which may explain the different neurobehavioral changes among the groups in the present study. Circulating IL-6 has also been reported to mediate inflammatory damage in the lungs after acute kidney injury in mice [42], supporting its role in triggering secondary ALI induced by distant organ damage. Additionally, we found that the blood level of IFN-γ was inversely related to SAH-induced ALI and the therapeutic effects of DEX. Despite the widely recognized role of IFN-γ in the stimulation of inflammatory responses to pathogens, its activity in sterile inflammation remains controversial [43]. Indeed, its blood level has shown to decrease in some pathological states, such as trauma and hepatitis [44,45], which is consistent with the current results. Decreased IFN-γ levels may contribute to a high incidence of pneumonia in patients with brain injury by creating an environment in which infectious pathogens thrive [46]. The expression of IFN-γ by immune cells in patients with viral respiratory infection and interstitial lung disease was lower in severe cases than in those with a moderate form of the disease [47]. In addition, a decrease in the blood IFN-γ level was shown to be a risk factor for lung fibrosis in this disease [48]. More sophisticated exploration of the role of circulating IFN-γ in the pathophysiology of ALI induced by SAH is needed.
The current study has several limitations. First, we did not proceed into molecular studies on the SAH+DEX50 group, since the high dose DEX was not protective against the ALI. Information on the autophagy and inflammatory signaling pathway could have helped in the understanding of no benefit and confirming the potential toxicity of the high dose, which needs future investigation. Second, although the role of autophagy in SAH-induced ALI was assessed using the lung expression of essential regulators of autophagy, we did not confirm this role by applying inhibitors or activators, which may not permit a definite conclusion. Nevertheless, the severity of lung damage and pulmonary edema was well correlated with autophagy activity, which may support the current assertion. Third, we did not assess alterations in lung mechanics and oxygenation function, which are important in defining ALI from a clinical perspective on SAH management. Fourth, mechanical ventilation was performed in all animals in our study in light of actual clinical situations, in which the majority of SAH patients needed ventilatory support. This could have contributed to the comparable protein expression levels of LC3B in the sham and SAH groups, although the severity of ALI was significantly different between the two groups. The presence of additional groups of sham and SAH without mechanical ventilation in the experimental protocol might have better clarified the molecular mechanisms underlying each of the SAH-induced ALI and VILI, as well as the impact of their combination, which could have provided superior information to improve the management of SAH patients regardless of ventilatory support. Fifth, the association of autophagy with the other specified pathophysiology of brain-lung crosstalk, such as oxidative stress or sympathetic overdrive, was not assessed in the current study, for which further evaluation is needed.
Overall, we demonstrated in this study that autophagic flux and the TLR-dependent inflammatory signaling pathway participate in ALI induced by SAH. We also proved that systemic inflammation, one of the theoretical mechanisms of brain-lung crosstalk, mediates SAH-induced ALI. These molecular findings might be potential therapeutic targets, as each of them was attenuated by the administration of DEX within 24 h after SAH. Prompt clinical research in patients with SAH to define the effects of DEX is warranted to support the current results.
Notes
Funding
This study was supported by a National Research Foundation of Korea grant (NRF 2017R1C1B5016266) funded by the Ministry of Science, ICT, & Future Planning, South Korea.
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Author Contributions
Dong Woo Han (Conceptualization; Investigation; Methodology; Project administration)
Ju Eun Oh (Conceptualization; Formal analysis; Methodology)
Beom Jin Lim (Formal analysis; Investigation)
Yeonseung Han (Data curation; Investigation)
Young Song (Formal analysis; Funding acquisition; Visualization; Writing – original draft)