Introduction
Mepivacaine, an intermediate-acting aminoamide local anesthetic that has a rapid onset time and a relatively low risk of toxicity, has been used for regional anesthesia, particularly in patients who have compromised cardiac function [1]. Mepivacaine causes the intrinsic vasoconstriction of blood vessels or attenuated blood flow both in vivo and in vitro [2,3,4,5,6,7,8,9]. The intrinsic vasoconstriction induced by local anesthetics seems to be partially correlated with the duration of analgesia the anesthetics produce [10,11]. Decreased perineural blood supply, which is the result of intrinsic contraction evoked by local anesthetic, may relate to decreased local anesthetic clearance [10,11]. Vasoconstriction is regulated by both calcium-dependent and calcium-sensitization mechanisms [12]. An increased intracellular calcium concentration ([Ca2+]i) due to either calcium influx from extracellular fluid or calcium released from the sarcoplasmic reticulum contributes to the calcium-dependent mechanism responsible for vascular smooth muscle contraction [12]. Mepivacaine-induced contraction seems to be inhibited by endothelial nitric oxide, which is stimulated by endothelial [Ca2+]i increment [7,13]. Mepivacaine-evoked vasoconstriction involves increased calcium sensitivity, which is evoked by calcium influx from extracellular space [9]. However, the mechanism responsible for the [Ca2+]i increase associated with mepivacaine-evoked vasoconstriction remains to be determined. Therefore, the objective of this in vitro study was to examine the mechanism (i.e., either calcium influx or calcium release from sarcoplasmic reticulum) that is responsible for the [Ca2+]i increase, which contributes to the mepivacaine-evoked vasoconstriction of isolated rat aortas without endothelium.
Materials and Methods
Gyeongsang National University approved all experimental procedures and protocols. All experimental procedures were done in accordance with the Guide for the Care and Use of Laboratory Animals provided by the National Academy of Sciences.
Preparation of aortic rings for tension measurement
We prepared aortic rings for isometric tension measurement as previously described [14]. Anesthesia of male Sprague Dawley rats weighing 250-300 g was induced by intramuscular injection of Zoletil 50 (tiletamine HCl 125 mg and zolazepam base 125 mg/5 ml; 15 mg/kg; Virbac Laboratories, Carros, France). We dissected descending thoracic aorta, and removed surrounding connective tissues and fat under microscope in a Krebs solution bath. Krebs solution was composed of 118 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 2.4 mM CaCl2, 25 mM NaHCO3, and 11 mM glucose. The aorta was cut into 2.5-mm-length rings that were suspended on Grass isometric transducers (FT-03, Grass Technologies, Warwick RI, USA) under 3.0 g resting tension in a 10 ml Krebs bath at 37Ōäā, and aerated continuously with 95% O2 and 5% CO2 to persist pH within the range of 7.35-7.45. We used 120 min for equilibrium of rings and changed the bathing solution every 30 min. We removed endothelium from aortic rings using a 25-gauge needle. When phenylephrine (10-8 M) produced a sustained contraction, we added acetylcholine (10-5 M) into organ bath, and less than 10% acetylcholine-induced relaxation was regarded as endothelial denudation. The magnitude of contraction evoked by isotonic 60 mM potassium chloride (KCl) was measured for all aortic rings and regarded as the reference value (100%) for comparing contractile responses induced by mepivacaine. After washing out the KCl from the organ bath with fresh Krebs solution and reaching to the recovery of the baseline resting tension, mepivacaine-evoked dose-response curves were obtained (detailed below). The Krebs solution also included the nitric oxide synthase inhibitor Nw-nitro-L-arginine methyl ester (L-NAME, 10-4 M) to prevent the nitric oxide release due to remaining residual endothelium [15,16].
Experimental protocols
We assessed cumulative mepivacaine dose (10-5 to 10-2 M)-response curves in aortas without endothelium pretreated with or without the following inhibitors: voltage-operated calcium channel (VOCC) inhibitor nifedipine (10-8, 10-7 and 10-6 M), inositol 1,4,5-trisphosphate (IP3) receptor blocker 2-aminoethoxydiphenylborate (2-APB: 5 ├Ś 10-6, 10-5 and 5 ├Ś 10-5 M) and store-operated calcium channel (SOCC) blocker gadolinium chloride hexahydrate (Gd3+: 10-6 and 5 ├Ś 10-6 M) [16,17,18,19]. We added the above-mentioned inhibitors into the organ bath 20 min prior to the addition of mepivacaine into organ bath. Increasing concentrations of mepivacaine were added to the organ bath after the low concentration caused a stable and sustained contraction for 5 min. In addition, we investigated the effect of calcium-free and low-calcium concentration solutions on mepivacaine-induced vasoconstriction in the aortas without endothelium. The aortas were incubated with either low calcium (1 mM) containing normal Krebs solution or Krebs solution without calcium containing 2 mM ethylene glycol-bis (╬▓-aminoethyl ether)-N,N,N',N',-tetraacetic acid for 20 min prior to the addition of mepivacaine [16].
Fura-2 loading and measurement of [Ca2+]i
Preparation for fura-2 loading and measurement of [Ca2+]i were performed as described previously [9,20]. Anesthesia of male Sprague-Dawley rats (250-300 g) was induced through an intraperitoneal administration of pentobarbital sodium (50 mg/ml) and bleeding from the common carotid artery was induced. We dissected the descending thoracic aorta as described in the tension measurement section. Eighteen rats were used in this protocol. [Ca2+]i was measured according to the method described by previous researches using the fluorescent Ca2+ indicator fura-2 [9,20]. We exposed muscle strips to the acetoxymethyl ester of fura-2 (fura-2/AM, 10 ┬ĄM) with 0.02% cremophor EL for 5-6 h at room temperature [9,20]. After loading, we washed muscle strips with a physiological salt solution at 37Ōäā for 20 min to remove uncleaved fura-2/AM and muscle strips were held horizontally in a temperature-controlled, 7 ml organ bath. The muscle strips were illuminated alternately (48 Hz) at two excitation wavelengths (340 and 380 nm). The level of 500-nm fluorescence (F340 and F380) was detected using a fluorometer (CAF-100; Jasco, Tokyo, Japan). The ratio of F340 to F380 (F340/F380) was regarded as an amount of [Ca2+]i. Absolute Ca2+ concentration was not measured in our experiment because the dissociation constant of the fluorescence indicator for Ca2+ in cytosol may differ from that obtained in vitro [21]. Therefore, the ratios of F340/F380 obtained in resting and 60 mM KCl-stimulated aortic muscle were regarded as 0% and 100%, respectively. Ratios of F340/F380 were detected with a PowerLab/400 using a chart program (AD Instruments, Colorado Springs, CO, USA). Muscle strips were placed under an initial 3.0 g resting tension. All strips that came from the same animal were used in different experimental protocols. Mepivacaine-induced dose (3 ├Ś 10-5 to 3 ├Ś 10-3 M)-[Ca2+]i curves were obtained in muscle strips with or without following inhibitors: verapamil (10-7 and 10-6 M), nifedipine (10-8 and 10-7 M), 2-APB (5 ├Ś 10-6, 10-5 and 5 ├Ś 10-5 M) and Gd3+ (10-6 and 5 ├Ś 10-6 M) [16,17,18,19]. These inhibitors were added to the organ bath 15 min prior to the addition of mepivacaine and remained until the end of experiment. In addition, we investigated the effect of either low calcium (1 mM) containing Krebs solution or Krebs solution without calcium on mepivacaine-evoked concentration-[Ca2+]i curves to determine whether the mepivacaine-induced [Ca2+]i increase was dependent on calcium influx from extracellular fluid [16].
Drug
All drugs were commercially available. Verapamil, nifedipine, L-NAME and Gd3+ were obtained from Sigma Chemical Co. (St Louis, MO, USA), 2-APB from Calbiochem (San Diego, CA, USA), and fura-2/AM (a calcium indicator) from Molecular Probes (Eugene, OR, USA). Mepivacaine was donated by Hana Pharmaceutical Co., Ltd. (Hwaseong, Korea). All concentrations are shown as the final molar concentration in the organ chamber. Nifedipine and 2-APB were dissolved in dimethyl sulfoxide (DMSO; final organ bath concentration: 0.1% DMSO). Unless otherwise stated, we dissolved and diluted all other drugs in distilled water.
Data analysis
Data are shown as the means ┬▒ SD. Vasoconstrictions evoked by mepivacaine are expressed as percentages of the maximum contraction evoked by isotonic 60 mM KCl. The [Ca2+]i changes induced by mepivacaine are shown as percentages of the maximum [Ca2+]i increment evoked d by isotonic 60 mM KCl. Vasoconstriction or [Ca2+]i in response to each mepivacaine concentration were compared using repeated measures analysis of variance (ANOVA) followed by the Bonferroni post-test. Regarding the effects of the inhibitor on the contraction and mepivacaine-induced [Ca2+]i increase, the statistical analysis was analyzed using two-way repeated measure ANOVA followed by Bonferroni's multiple comparison test. Statistical analysis was performed with GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego, CA, USA). P values less than 0.05 were considered significant.
Results
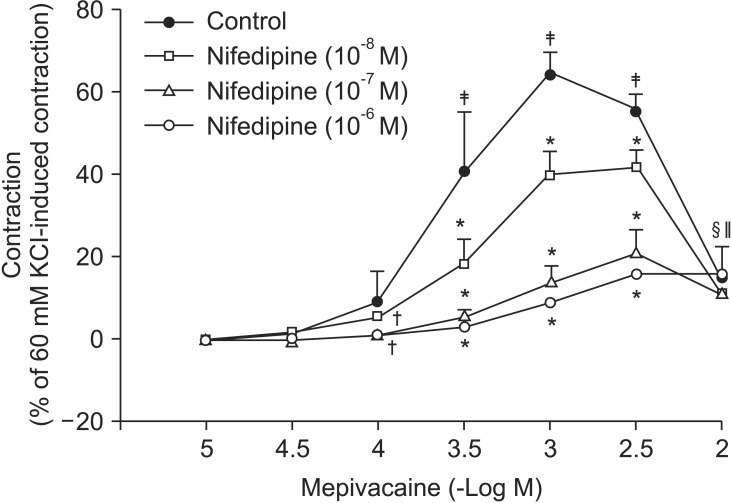
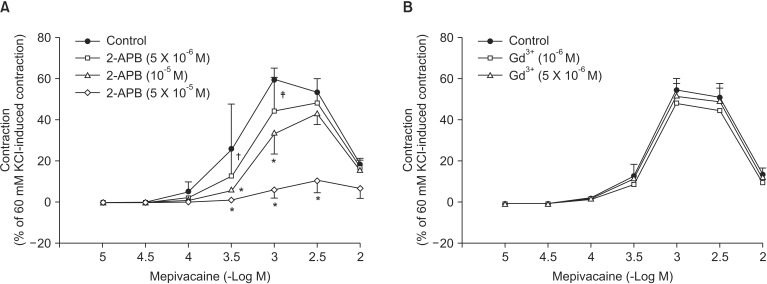
Mepivacaine (3 ├Ś 10-4 to 3 ├Ś 10-3 M) caused vasoconstriction (P < 0.001 versus 10-5 M mepivacaine) followed by attenuated vasoconstriction at 10-2 M mepivacaine (P < 0.01 versus 10-5 M mepivacaine; P < 0.001 versus 3 ├Ś 10-3 M mepivacaine) (Fig. 1). VOCC inhibitor nifedipine (10-8, 10-7 and 10-6 M) inhibited mepivacaine-evoked vasoconstriction (P < 0.05 versus control at 10-4 M mepivacaine, P < 0.001 versus control at 3 ├Ś 10-4 to 3 ├Ś 10-3 M mepivacaine, Fig. 1). IP3 receptor blocker 2-APB (5 ├Ś 10-6, 10-5 and 5 ├Ś 10-5 M) attenuated mepivacaine-evoked vasoconstriction (P < 0.05 versus control at 3 ├Ś 10-4 and 10-3 M mepivacaine) (Fig. 2A). However, the SOCC blocker Gd3+ had no effect on mepivacaine-evoked vasoconstriction (Fig. 2B). A low calcium level (1 mM) significantly decreased the mepivacaine-evoked vasoconstriction (P < 0.05 versus control at 3 ├Ś 10-4 M and P < 0.001 versus control at 10-3 M mepivacaine, Fig. 3), and Krebs solution without calcium almost abolished mepivacaineevoked vasoconstriction (P < 0.001 versus control at 3 ├Ś 10-4 to 3 ├Ś 10-3 M mepivacaine, Fig. 3).
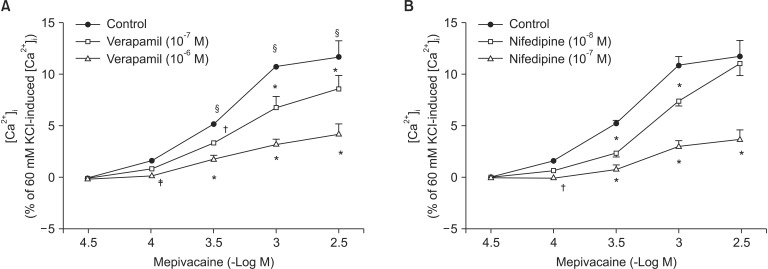
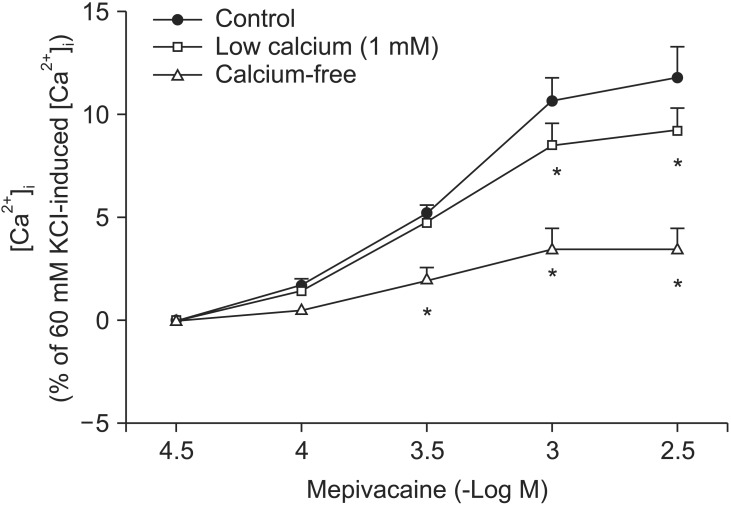
Mepivacaine (3 ├Ś 10-4 to 3 ├Ś 10-3 M) increased [Ca2+]i (P < 0.001 versus 3 ├Ś 10-5 M mepivacaine, Fig. 4A). The VOCC inhibitors verapamil (10-7 and 10-6 M) and nifedipine (10-8 and 10-7 M) attenuated the mepivacaine-induced [Ca2+]i increase (P < 0.01 versus control at 3 ├Ś 10-4 and 10-3 M mepivacaine, Fig. 4A and B). The IP3 receptor blocker 2-APB (10-5 and 5 ├Ś 10-5 M) inhibited the mepivacaine-evoked [Ca2+]i increase (10-5 M 2-APB: P < 0.001 versus control at 10-3 M mepivacaine, P < 0.05 versus control at 3 ├Ś 10-3 M mepivacaine; 5 ├Ś 10-5 M 2-APB: P < 0.001 versus control at 3 ├Ś 10-4 to 3 ├Ś 10-3 M mepivacaine, Fig. 5A), whereas the SOCC inhibitor Gd3+ had no effect on the mepivacaine-evoked [Ca2+]i increment (Fig. 5B). Low calcium concentration (1 mM) attenuated the mepivacaine-induced [Ca2+]i increase (P < 0.001 versus control at 10-3 and 3 ├Ś 10-3 M mepivacaine, Fig. 6). Calcium-free Krebs solution strongly inhibited the mepivacaine-induced [Ca2+]i increase (P < 0.001 versus control at 3 ├Ś 10-4 to 3 ├Ś 10-3 M mepivacaine, Fig. 6).
Discussion
Taking the results together, this is first study to suggest that the mepivacaine-induced [Ca2+]i increase associated with mepivacaine-induced contraction seems to be mainly dependent on the calcium influx from extracellular space. The main findings of current study were as follows: 1) nifedipine and 2-APB inhibited mepivacaine-evoked vasoconstriction; 2) low calcium level inhibited mepivacaine-evoked vasoconstriction, and Krebs solution without calcium almost abolished mepivacaine-evoked vasoconstriction; 3) verapamil, nifedipine, 2-APB and low calcium level inhibited the mepivacaine-induced [Ca2+]i increase; and 4) calcium-free solution strongly attenuated the mepivacaine-induced [Ca2+]i increase.
Vasoconstriction induced by a receptor-mediated contractile agonist (example: phenylephrine) involves [Ca2+]i increase via both calcium influx from extracellular fluid and calcium released from the sarcoplasmic reticulum [22]. Calcium influx that contributes to phenylephrine-evoked vasoconstriction is mediated by VOCC and receptor-mediated calcium channels [22,23,24]. IP3 receptor-induced calcium release from sarcoplasmic reticulum causes transient initial vasoconstriction evoked by phenylephrine [22]. Consistent with the results of previous reports, VOCC inhibitor nifedipine inhibited mepivacaine-induced contraction and the highest concentration (10-6 M) of nifedipine almost inhibited mepivacaine-evoked vasoconstriction, suggesting that mepivacaine-evoked vasoconstriction involves activation of VOCC [7,8,9]. 2-APB, a blocker of IP3 receptor associated with calcium release from the sarcoplasmic reticulum, inhibited mepivacaine-evoked vasoconstriction, suggesting that mepivacaine-evoked vasoconstriction involves IP3 receptor-induced calcium release from the sarcoplasmic reticulum [17,18]. However, 2-APB is a inhibitor not only of IP3 receptor but also of SOCC [17,18,25,26,27]. Therefore, the inhibitory effect of 2-APB on mepivacaine-evoked vasoconstriction may be associated with the blockade of SOCC rather than that of the IP3 receptor. To rule out 2-APB-induced SOCC inhibition as a contributor to the inhibition of mepivacaine-induced contraction, we evaluated the effect of SOCC inhibitor Gd3+ on mepivacaine-evoked vasoconstriction. Because Gd3+ had no effect on mepivacaine-evoked vasoconstriction, the inhibitory effect of 2-ABP on mepivacaineevoked vasoconstriction appears to be due to inhibition of IP3 receptor. Furthermore, a low calcium concentration (1 mM) inhibited mepivacaine-evoked vasoconstriction, and Krebs solution without calcium almost abolished the effect. Taken together, these results suggest that mepivacaine-evoked vasoconstriction seems to be mediated mainly by calcium influx via VOCC and partially by calcium released from the sarcoplasmic reticulum. Further research to investigate the mechanism that is responsible for high-dose (10-3 ├Ś 3 and 10-2 M) mepivacaine-induced attenuated vasoconstriction is thus required.
To confirm that mepivacaine-evoked vasoconstriction is mainly dependent on calcium influx, as observed in the isometric tension experiment, we assessed the effect of various inhibitors on mepivacaine-evoked [Ca2+]i increase using aortic strips loaded with fura-2. Mepivacaine increased [Ca2+]i (Fig. 4A), which contributes to the mepivacaine-evoked contraction. Consistent with the results from isometric tension measurements in the current study, nifedipine and verapamil attenuated the mepivacaine-induced [Ca2+]i increase. In addition, 2-APB (10-5 and 5 ├Ś 10-5 M) attenuated the mepivacaine-induced [Ca2+]i increase, whereas Gd3+ had no effect. Taken together, the mepivacaine (3 ├Ś 10-4 to 3 ├Ś 10-3 M)-induced [Ca2+]i increment seems to be mediated by both calcium influx through VOCC and calcium released from the sarcoplasmic reticulum. Furthermore, because a low calcium concentration attenuated the mepivacaine-evoked [Ca2+]i increment and Krebs solution without calcium strongly inhibited the mepivacaine-evoked [Ca2+]i increment, the mepivacaine-evoked [Ca2+]i increment appears to be primarily dependent on calcium influx through VOCC. Further research regarding the upper cellular signal pathway responsible for the mepivacaine-induced [Ca2+]i increase is needed to elucidate detailed signal pathway. In agreement with a previous study that enhanced calcium sensitivity evoked by Rho-kinase and protein kinase C contributes to mepivacaine-induced contraction, maximum contraction induced by mepivacaine was 65 ┬▒ 5% of 60 mM KCl-induced contraction (Fig. 1), whereas the maximum [Ca2+]i induced by mepivacaine was 12 ┬▒ 5% of 60 mM KCl-induced maximum [Ca2+]i (Fig. 4A), suggesting that mepivacaineevoked vasoconstriction is mediated both mainly by calcium sensitization and partially by increment of [Ca2+]i [9].
The clinical relevance of mepivacaine-induced contraction mainly mediated by an increment in [Ca2+]i via calcium influx must be tempered by the following factors. First, we used the aorta, which has a role as conduit vessel, whereas small resistance arterioles regulate organ blood flow such as perineural blood flow [28]. Second, endothelial nitric oxide inhibits contraction evoked by mepivacaine [7]. In addition, the shear stress-induced nitric oxide release by blood flow in vivo would inhibit mepivacaine-induced vasoconstriction [29]. Considering these factors, nitric oxide release in an in vivo state attenuates mepivacaine-induced contraction observed in this in vitro study [7,29]. Despite these limitations, the pale skin color, inhibited capillary blood flow and reactive hyperemia, and reduced cutaneous blood flow observed during mepivacaine-evoked analgesia in previous in vivo researches and intermediate-acting period of mepivacaine may be due to mepivacaine-induced vasoconstriction mediated by an increase in [Ca2+]i [2,4,5,6,30]. In addition, mepivacaine-induced contraction may be attenuated in a patient who takes a calcium channel blocker, leading to a short duration of mepivacaine-evoked analgesia compared with a person who does not take a calcium channel blocker. The clinically available concentrations of mepivacaine are between 0.5 and 2%, which approximately corresponds to 1.725 ├Ś 10-2 to 7 ├Ś 10-2 M. This concentration produced vasodilation in the current study. However, the clinically available concentrations of mepivacaine produce reduced blood flow and vasoconstriction in vivo [4,5,6]. This difference in the mepivacaine-induced vascular response between the current study and previous researches may be due both to differences in blood vessels (capillary versus aorta), species (human versus rat) and experimental methods (in vivo versus in vitro) and to the dilution of infiltrated mepivacaine using interstitial fluid in an in vivo state. When mepivacaine is locally infiltrated, the local tissue level of mepivacaine may be lower than the commercially available concentration of mepivacaine because locally applied mepivacaine is diluted by interstitial fluid and 77.5% of mepivacaine is bound to plasma protein [1].
Taken together, these results suggest that the mepivacaineevoked [Ca2+]i increment that contributes to mepivacaine-induced contraction appears to be mediated both primarily by calcium influx via VOCCs and partially by calcium released from the sarcoplasmic reticulum in isolated endothelium-denuded rat aortas.